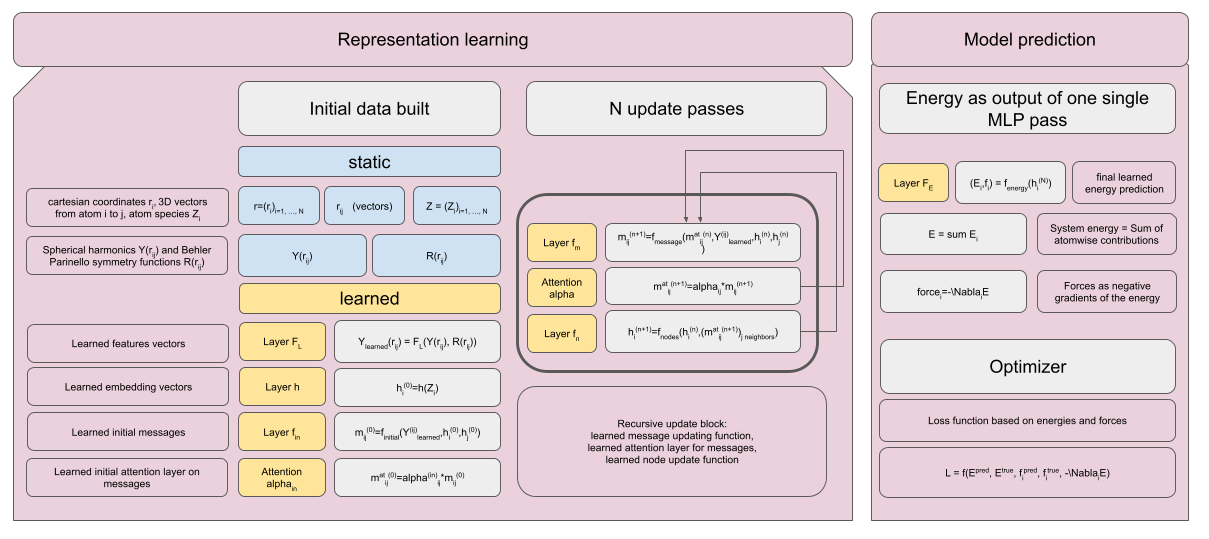
Network Architecture
The goal of the model is to predict atom-wise energy contributions to a chemical system (e.g. one single molecule). That is, the model needs to learn a suitable representation for each atom in the system. Initial representation will be divided in two key parts: Radial and directional features. Radial features will be based on pairwise distances. Directional features will be constructed using vector-based descriptors (this is the part where we need the neighbor list). For both types of features, we can use some fixed initial descriptors such as symmetry functions, spherical harmonics, Bessel functions etc., that are then passed through a learnable MLP. The node vectors will be updated iteratively (-> representation learning).
Some succesful architectures, such as MACE[1] or NequIP[2], explicitly ensure equivariance by using tensor representations instead of vectors. ANI[3] uses triplets and fixed angular features to account for 3D-arrangements. However, my goal is to propose a less accurate but hopefully faster architecture. Therefore, I plan to only use a directional edge representation (no atom triplets and no higher-order tensors). This design retains directional awareness at hopefully lower computational cost compared to real equivariant networks. This will surely reduce the network’s potential accuracy, but also hopefully reduce computation times.
Subsections
[1] MACE (https://doi.org/10.48550/arXiv.2206.07697)
[2] NequIP (https://www.nature.com/articles/s41467-022-29939-5)
[3] ANI (doi.org/10.1039/C6SC05720A)